Intertek supports Medical Device manufacturers in meeting all Safety and Quality requirements around the world
Simplifying the complex world of medical device compliance requires a trusted partner with unparalleled expertise. Intertek is committed to helping you bring innovative and high-quality medical devices to market efficiently and confidently.
We offer comprehensive medical device assurance, testing and certification solutions designed to meet stringent regulatory requirements, including FDA guidance, 60601-1 standards, and other global standards. Our services ensure your devices not only comply with the latest safety and performance regulations but also gain faster market entry to every region around the world.
From electrical safety and EMC to cybersecurity, biocompatibility and beyond, our experts are well-versed in the challenges of today’s medical device regulatory and compliance landscape. Whether you’re developing never-before-seen technologies or improving existing FDA-approved medical devices, Intertek provides the assurance you need to succeed.
Intertek's Solutions for Medical Devices
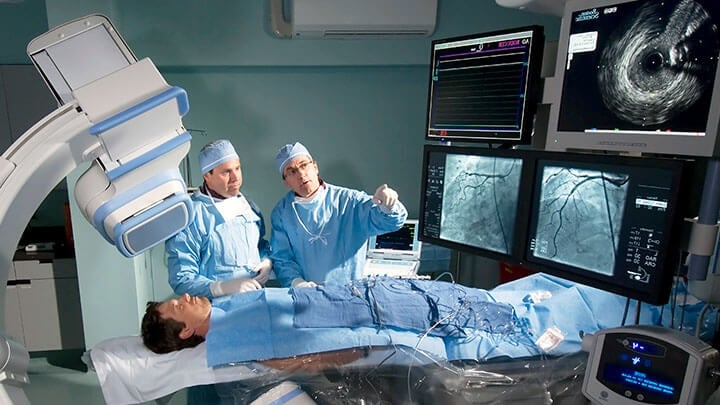
Compliance Solutions for Safety and EMC Standards
Solutions to meet state-of-the-art standards on safety and electromagnetic compatibility (EMC), including IEC 60601-1, 60601-1-2, Home Healthcare, medical imaging equipment, IEC 61010, Implantable medical devices, SPE-3000 and more.
Global Regulatory Requirements
Critical information to effectively navigate Global Regulatory Requirements such as CB scheme, FDA requirements, ASCA, NMPA, Japan, Brazil, and more.

Connected and Digital Technologies
The latest information and expert resources regarding digital and connected medical technologies such as Software as a Medical Device (SaMD), Wireless devices, AI-Enabled and Machine Learning devices.

Performance Related Testing Solutions
Critical performance testing services to test and validate the quality of your Medical Devices, including battery testing, Reese’s Law compliance, testing for home healthcare environments, biocompatibility, reliability, benchmarking and more.

Sustainability Solutions and Services
Access to Assuris, our global network of scientists, engineers, and regulatory specialists providing support to navigate complex scientific, regulatory, environmental, health, safety, and quality challenges throughout the value chain.
Medical Devices Auditing and Certification Services
Auditing, performance, benchmarking and supply chain solutions to deliver insights and quality management into every aspect of your operations.
Medical Device Testing and Scientific Support Services
Unrivaled expertise in Medical device materials testing, chemical analysis and scientific support.
Clinical Studies
Cosmetic clinical trial expertise in clinical safety and efficacy testing of cosmetics, beauty products and devices to support claims and demonstrate conformity with regulations.

Related Links
Featured on #YoullBeAmazed

See this post on Facebook, LinkedIn, or learn more about our You'll Be Amazed campaign.

Medical Devices Q&A: Ensuring a Smooth Path to Certification
Ensure a seamless certification process for your medical device with this exclusive Q&A to discover actionable advice on avoiding common pitfalls, streamlining documentation, and meeting regulatory standards efficiently.
Common Medical Device Products
- Diagnostic Equipment
- Surgical Instruments
- Therapeutic Equipment
- Monitoring Devices
- Implantable Medical Devices
- Dental Devices
- Assistive Devices
- In vitro Diagnostic Devices
- Health Monitoring Devices
- Home Healthcare Devices
- Fitness Devices
- Laboratory Equipment
24 Jan 2025
Navigating the FDA's New Draft Guidance on Chemical Analysis for Medical Devices
What You Need to Know

Knowledge Center
Download the latest information from our medical device compliance experts.
Six Compliance-Related Questions About Connected Home Healthcare Devices
Accelerated Stress Testing for Medical Devices
Machine Learning and Artificial Intelligence (AI) in Medical Devices: Webinar | Fact Sheet
Chemical Analysis for Biocompatibility Assessment of Medical Devices Draft Guidance for Industry
ENERGY STAR® Requirements for Medical Imaging Equipment
Creation of IEC 60601-1 4th Edition
IEC 60601-1-2 Ed. 4.1 Overview of Requirements
Medical OEM Wireless Coexistence Testing
Biocompatibility Risk Assessment and Evaluation Plans
For more expert papers, recordings, and presentations, visit our Medical Resources hub.


Follow Us For More!
We’re always adding new content and looking for ways to help you simplify the regulatory and compliance process for medical devices.
** The Intertek legal entities that provide medical device management system certification services (including ISO 13485 and MDSAP) and Notified Body services (MDR 2017/745 and MDD 93/42/EEC) do not provide any consulting services. Clients who have used other Intertek legal entities’ consulting services are not eligible to receive management system certification services or Notified Body services from Intertek.
